Occasionally online I will get criticized for past statements I’ve made about my stance on truth in science. This criticism is usually from people who have a very simple view that to do science one must seek out truth in nature. There are, of course, many ways to do science and to be a scientist and it does not require a commitment to a particular picture of a mind-independent reality or to science’s ability to accurately capture a mind-independent reality. My position is that I don’t set my sights on capturing these truths about a mind-independent reality when doing science, I don’t think many other scientists actually do either. In physics the position of “shut up and calculate” is common, famous physicists like Ernst Mach, Werner Heisenberg, and arguably Niels Bohr were anti-realists of some kind. Heisenberg once said
[physicists] would prefer to come back to the idea of an objective real world whose smallest parts exist objectively in the same sense as stones or trees exist independently of whether we observe them. This however is impossible…1
and physics is on far more solid ground than fields like biology. I’m also a bit skeptical of science’s ability to give much access to this kind of truth in the grand scheme of things, especially for some fields of science.
An important clarification is that when I, and most others, say I’m skeptical of the ability of science to truthfully describe an underlying mind-independent reality this doesn’t mean I don’t aim for rigor and accuracy in my science, it just means my goal is to provide careful, reliable, and rigorous analysis and conclusions that can sufficiently describe/explain my observed data and potentially make novel predictions. Just because my theories and hypotheses can do this does not mean they are literally true or properly describing a mind-independent reality. It also doesn’t necessarily mean I think there is no mind-independent reality, simply that science may not always be able to actually describe it. This position puts me in the scientific anti-realist2 camp, somewhat close to Constructive Empiricism3 as described by the philosopher Bas Van Fraassen, though I have neither the time nor the motivation to pick a specific flavor of anti-realism. Generally, I am simply skeptical that our theories provide a True picture of the nature of reality and prefer to restrict my goals to something with more epistemic modesty. Much of this skepticism and anti-realist sentiment, especially about my own field, makes even more sense because traditionally even scientific realists who believe science can and should truthfully describe a mind-independent reality intend to restrict the domain of theories and fields relevant to a realist commitment to mature sciences, where mature is normally referring to well and long established fields. Typically mature fields are expected to have a high degree of matching between theory and data. My field of genomics, and arguable biology as a whole, are far from a mature science. As much as I love genomics I have to recognize that the field is full of assumptions on top of assumptions and models on top of models that idealize away parts of the biological processes we have good reason to believe exist in a way that keeps me up at night. Sharing these concerns will hopefully inspire some skepticism and caution in others when reading genomic studies as well.
The Genome is just a Map
The first watering down of reality is the way we study genomes. We have some idea that genomes, consisting of protein bound DNA in bundles of chromosomes, are three dimensional. Chromosomes bend and contort and regions that are not linearly close to each other or on the same chromosomes can come into contact and interact by these 3D conformations in important ways. Despite this, our representations of genomes are predominately a string of characters organized into separate chromosomes that don’t encode the higher level spatial information about genome organization. Technology is slowly allowing some analysis of these aspects and, for example, their importance in gene regulation4 but generally this information is not included in most genomic studies. Problems with representational ability of genomes doesn’t just end there. Genomes also commonly end up leaving much of the physical genetic material unsequenced due to complexity of the regions. To understand this we should briefly talk about how genomes are sequenced. The most common method involves taking strands of DNA, cutting them up into smaller pieces, sequencing those pieces into what are called reads, and then piecing together the genome by tiling reads based on regions of overlap
 .
.
One can do this guided by a “reference genome”
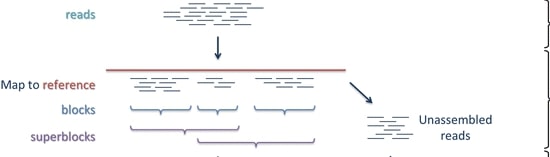
or without a reference, de novo.
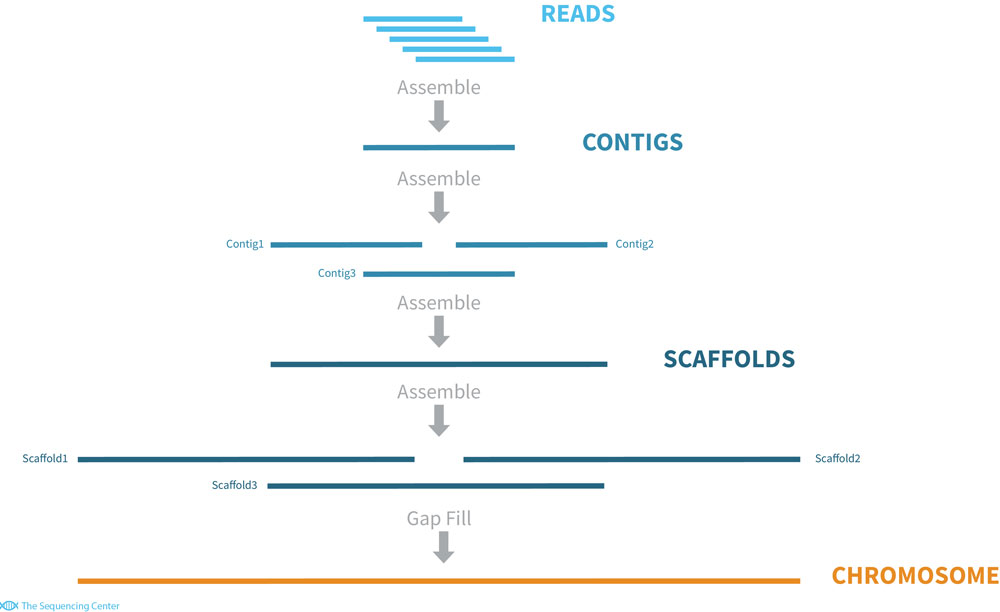 The difficulty is that the short reads of this method cannot cover highly repetitive or complex regions of the genome such as centromeres or where repetitive elements have invaded. This leaves gaps in genome assemblies and means there are parts of the physical genome not present in our representation. In many cases we miss out on hundreds or thousands of genes5, including some involved in adaptive traits. There have been many advancements in genome technology and more complete reference genomes are being made, but some organisms still have no or imperfect reference genomes and cannot easily have a high quality genome made. Often we are dealing with a substantially incomplete picture of genetic material for an organism.
The difficulty is that the short reads of this method cannot cover highly repetitive or complex regions of the genome such as centromeres or where repetitive elements have invaded. This leaves gaps in genome assemblies and means there are parts of the physical genome not present in our representation. In many cases we miss out on hundreds or thousands of genes5, including some involved in adaptive traits. There have been many advancements in genome technology and more complete reference genomes are being made, but some organisms still have no or imperfect reference genomes and cannot easily have a high quality genome made. Often we are dealing with a substantially incomplete picture of genetic material for an organism.
What happens when the map isn’t annotated well?
Much of the power of genomes comes from the fact that we use them to locate genes throughout the genome (structural annotation) and estimate their function (functional annotation). This process is called genome annotation. For the majority of genomes automated computational methods are used to annotate the genome based on trained statistical models that in part rely on genomes of closely related species^6. Unfortunately quality of genome annotation is affected by the quality of genomes, as poorly covered regions of genomes cannot be annotated very well. The result is many genome annotations contain errors and misannotations. Even worse, these errors tend to propagate as faulty annotations are used when annotating other genomes and there is no reliable way to update all genomes built upon an annotation when errors are found and fixed6. Bad genome annotations also have major implications for downstream analyses that try to identify genes that cause traits of interest and will result in the inability to identify relevant genes or produce false positives7. It’s because of this that genome annotations are usually updated frequently. These updates generally lead to improvements in annotation quality8. However this means that the analyses you do might be dependent on the annotation version you use and results might not replicate in updated annotations. Such occurrences have been observed for clinically relevant cases like lung cancer9.
One genome isn’t enough
Even ignoring the idealizations involved in reference genomes and genome annotation, we find ourselves in a poor situation. The idea of a reference genome is that the genome of a single genotype can capture the genomic diversity of an entire species. It’s assumed or hoped that this can sufficiently represent the diversity of the species in a way that won’t hinder investigation, but recently it has been shown this assumption doesn’t hold. Pan-genomes are constructed by sequencing multiple genotypes and meant to identify the “core genome” shared by all genotypes and the “dispensable genome” that is variable between genotypes10. So far, pan-genomes have been constructed for bacteria, plant and animal species. In some plants it’s been found that as much as 50% of the genes in the pan-genome are variable among different genotypes, in humans an additional ~300Mb of sequence was found. Essentially when we use a reference genome for diverse individuals we are missing out on tremendous amounts of sequence, potentially hundreds or thousands of genes, and complex aspects of genomes like transposable elements that are important for genomic and evolutionary analyses11. The implications are pretty clear. When you’re using a reference genome when analyzing a different genotype there are likely many aspects of the genome you are missing and your analyses are now confounded by the fact that there are genotype specific genomic features you are not able to capture.
Gene Expression
After these issues with genomes, we are faced with more idealizations in our efforts to model biological systems. RNAseq, or transcriptomic, attempts to measure and quantify gene expression for all expressed genes in a particular organ and species. It involves extracting RNA from studies and using a similar short-read sequencing process as used in genomic methods described above. Typically studies using RNAseq are interested in identifying genes that have significantly different expression under an experimental treatment or condition like having a disease vs not having a disease. Sometimes researchers also care about broad patterns of expression of genes, such as my work that was interested in how different parental genomes of hybrid species contribute to overall gene expression12. The first concern with the majority of studies looking at gene expression is that these studies involve a process that combines several distinct types of cells at once, obscuring important patterns in the data. We have good reason to believe that different types of cells have different expression patterns, red and white blood cells, epithelial cells, muscle cells, etc have different forms and engage in different processes and so will involve different genes or express them in different ways. Combining them together and only getting the average of all cell types in a given tissue loses tremendous amounts of information. It’s difficult to interpret results or extrapolate too far without more knowledge about cell-specific results. Single-Cell RNAseq is becoming more wide-spread but is technically challenging and not always feasible in all experimental contexts. This means RNAseq studies are inherently limited in how well they can model biological processes.
Beyond issues with bulking cell types together, the actual measuring and quantification of RNAseq data is not a trivial task. RNAseq is generally accurate, but there are many methods used to align RNAseq reads to a reference genome, methods to quantify transcript expression and to identify genes that are significantly differentially expressed between treatment groups13. These methods rely on different techniques, algorithms, and models and so do not give identical results14. Often it isn’t clear which method is best or which model makes more realistic assumptions so the use of tools is largely conventional and based on what is available at a given time. While these differences are not always large it still means that given certain contexts it is possible that some portion of results are driven by methods employed by the experimenter rather than biological signal we are wanting to capture.
Yet another potential issue arises with our analysis because typically when we perform these studies we sample a single time point. Unfortunately for us, many genes have a rhythmic expression that cycles throughout the day, also known as circadian rhythm. In plants and animals this circadian expression is widespread and affects many genes and physiological responses15,16. Oftentimes the expression difference between a gene at a different time point is larger than the difference in expression caused by an experimental treatment so ensuring time of sampling is the same for control and treatment groups is vital to remove confounding introduced by circadian expression. Additionally because circadian rhythm is related to stress responses, especially in plants, some patterns of gene expression to response can only properly be captured by looking at certain time points or changes in circadian expression patterns17. Again we may find ourselves missing vital information and ignoring important confounding issues because of logistical or methodological limitations of a study.
Lost in translation from lab to field
Unfortunately the problems do not stop with the methodological aspects, there are also may ways that experimental design and experimental conditions can confound experiments. For example, one study that looked at three plant genotypes grown in ten different labs supposedly following the same protocols found that lab environments greatly affected leaf phenotypes as well as metabolite composition18. Meaning many effects observed from experiments may be due to particularities of a particular lab’s growing condition and not a general phenomena. The problems don’t stop from interlaboratory variation though, it has also been observed that for many plant species, several traits vary highly whether they are grown in growth chambers, greenhouses, or in the field19. We may see a certain result in a growth chamber but it doesn’t occur in the field. Even worse we may identify a genetic association in the growth chamber and it may not hold in the field and vice-versa. We can even see the rank-ordering of genotypes change between environments, with the genotype with the highest trait value in growth chambers being the lowest in the field. Researchers may not always be able to perform trials in the growth chambers and the field or know all the relevant growth variables, meaning impacts of these potential confounds are always lurking in experiments.
Conclusion
Although I’ve been very cynical in this piece I do recognize the power of genomic analyses. The ability to interrogate biological phenomena at the genetic level with high levels of precision has been a huge aid to biological investigation, helping make advancements in agriculture and medicine. However, it is also a field that is constantly developing, we’re building knowledge while building methods to acquire knowledge and often we have not perfected or validated some of these methods. There are constant new findings about limitations of confounds to the approaches we take that can alter or reverse conclusions. The resolutions to these problems likely won’t occur during a researcher’s career and the models, hypotheses, and theories they come up with can easily be overturned due to methodological problems. At its core this is why I’m not heavily invested in discovering truth from my scientific investigations. If I were to try to meticulously account for every possible confounds that I list above, I would never complete my research! Some of the work I do, like phylogenetics where I’m modeling the past evolutionary relationships between species may not ever be provable since we don’t have a time machine and can rarely if ever externally validate our estimations. My advisor is always prone to remind me that phylogenetic trees are just hypotheses based on available data and using available methods. I’m happy to settle for just being able to explain my observed data with my model or theory and attempt to explore the accuracy and utility of that, perhaps making new predictions along the way. Considering the state of genomics and the limitations of what a single researcher can do it feels more responsible to constrain my goals and focus on more pragmatic and realistic outcomes.
-
Heisenberg W.: Physics and Philosophy. New York: Harper and Row 1958, page 129
-
https://plato.stanford.edu/entries/scientific-realism/#AntiFoilForScieReal
-
https://plato.stanford.edu/entries/constructive-empiricism