The spectacular diversity of life reflects a continual interplay between ecological opportunity and evolutionary innovation. But before selection can shape new forms and functions, variation must first be generated, and the genome determines not only how much variation arises, but what kinds of variation are possible. My research asks how large-scale changes to genome architecture alter the evolutionary potential of lineages. Why do lineages differ so much in how their genomes restructure over evolutionary time? How do these processes generate, constrain, and reorganize the variation available to selection? And when does genome restructuring create opportunities for evolutionary novelty? Because these same processes shape both the crops we depend on for food and fuel and the invaluable wild plant diversity that institutions like Kew work to conserve, understanding how genomes enable or constrain evolutionary change has important implications for agriculture, biodiversity, and conservation.
Why do genomes restructure differently across lineages?
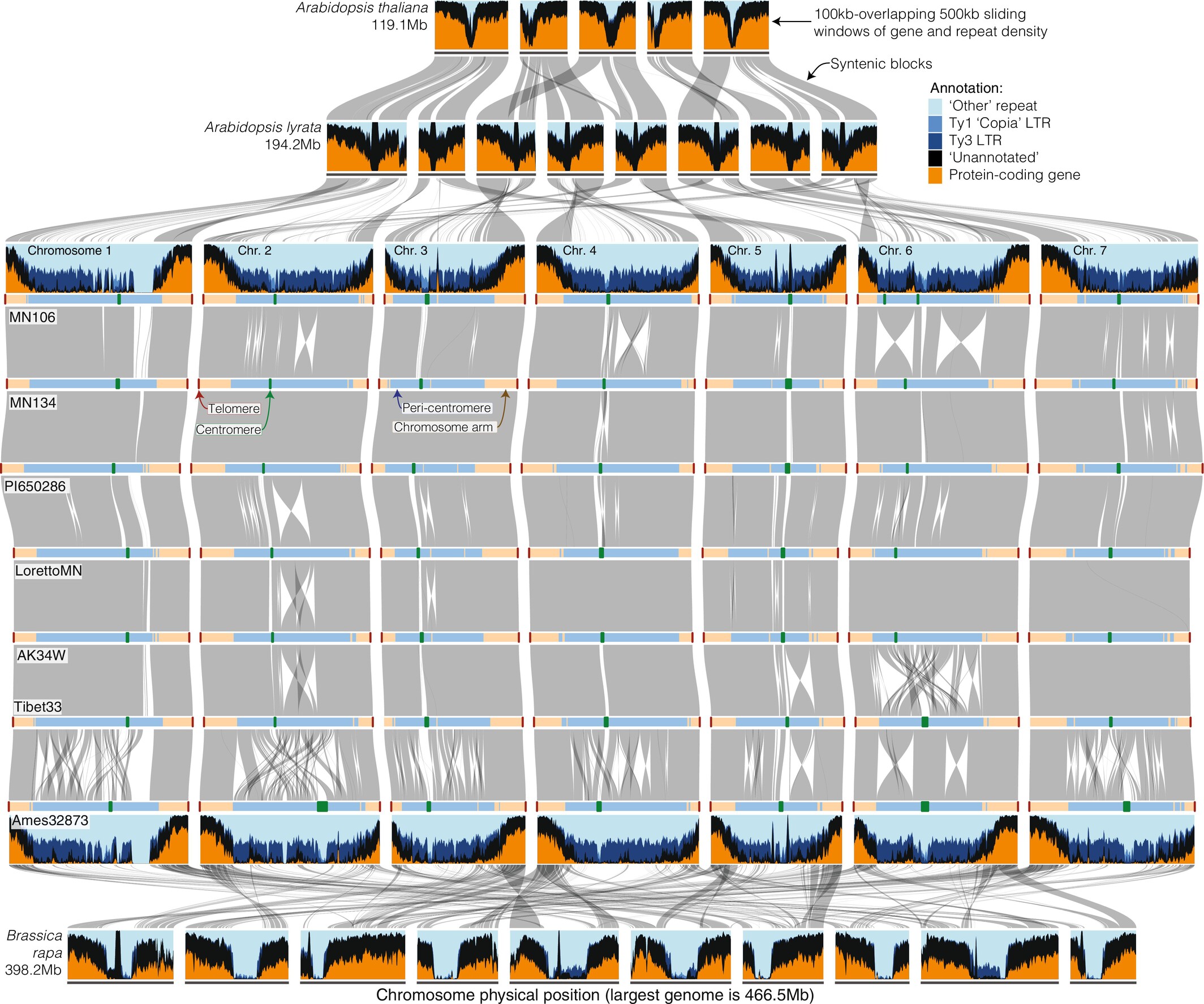
Ferns rarely lose chromosomes after whole-genome duplication, yet such losses are common in angiosperms. Gymnosperms almost never undergo whole-genome duplication at all, while angiosperms have done so again and again. And within the angiosperms, the rose family changes its chromosome number and structure far more slowly than the mustard family. Why genomic change differs so sharply across branches in the tree of life is still largely unresolved. My research seeks to understand what governs that difference, characterizing patterns of genomic variability and identifying the factors that promote or inhibit genome evolution. I’ve used resynthesized polyploids to show that inherited differences between parental genomes create expression imbalances in polyploid plants (Bird et al., 2021 New Phytologist), constructed species-level pangenomes to test whether whole-genome duplication elevates pangenomic variation (Bird et al., 2025 Genetics), and characterized how genome architecture itself shapes the magnitude and kind of genetic variation across the genome (Bird & Rifkin et al., 2026 New Phytologist). In my current work, I’m looking across land plants to understand how lineages return to a diploid state after whole-genome duplication, trying to disentangle how chromosome behavior and structure evolve and the functional consequences of that variation.
How is variation in the genome organized and reorganized?
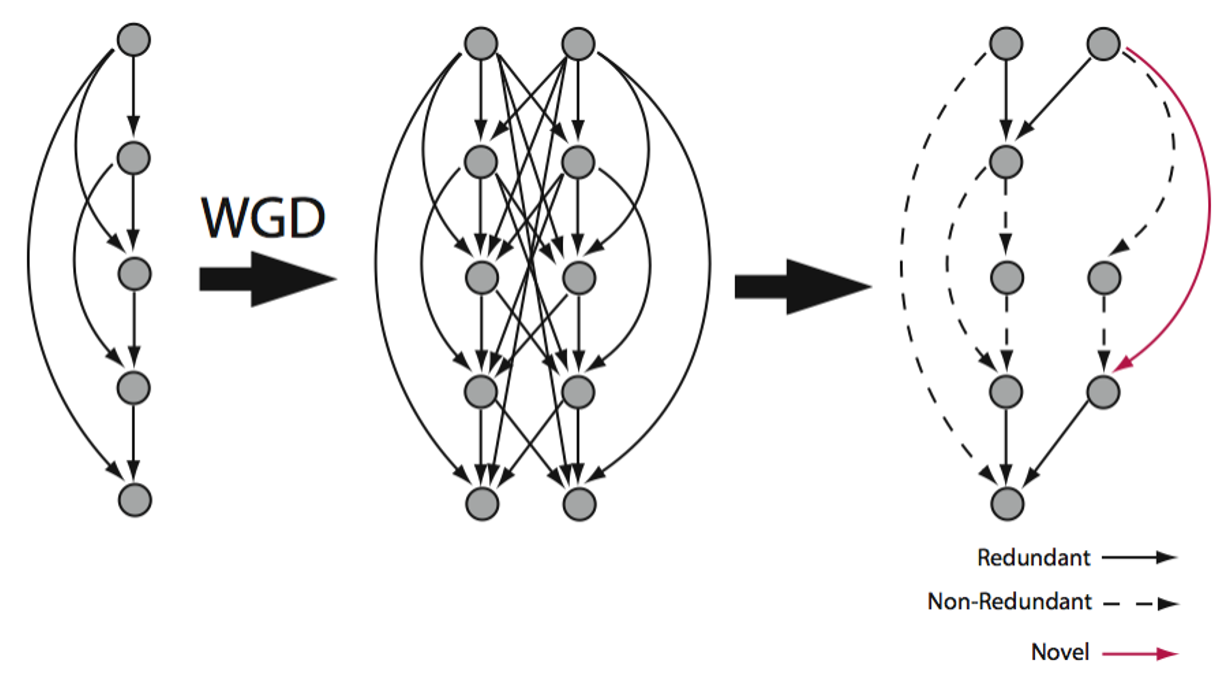
Genomes aren’t loose collections of independent genes; they’re coevolved systems of interacting parts, tuned by selection to work together within networks, pathways, and modules. My recent work is exploring how that organization actively shapes how genetic variation is distributed, structured, and buffered at the level of these coevolved units. In a pangenome of Arabidopsis thaliana, I’m finding that the dispensable genome is organized into coevolved functional modules, not a catalog of optional genes that vary independently across individuals. Recent work with resynthesized polyploids has found a parallel signature at the level of gene expression: functionally related genes are often maintained as duplicates by selection to preserve stoichiometric balance, and in doing so continue to buffer one another’s expression against genomic change. Both studies point to the same conclusion that the genome’s coevolved functional modules structure which combinations of variants are accessible and buffer the system against perturbation. When genomes are restructured by duplication, gene loss, or rearrangements, this reshapes not just individual genes but the coordinated units that selection ultimately sees. When a whole module is reorganized at once, the consequences can produce something entirely new.
When does restructuring produce novelty?
Ultimately, I want to know when system-level genomic change translates into new functions, new phenotypes, and new ecological possibilities. Metabolic pathways are an exceptionally clean model to study this, because their chemistry can be read almost directly from their genes. My work has focused on clarifying our understanding of the molecular changes that underlie metabolic innovation. In comparative work across the Brassicales on the glucosinolate pathway (responsible for the pungent chemistry behind mustard, wasabi, and horseradish), I found cases of novel chemotypes across species that arose not from gene-by-gene tinkering, but from rapid coordinated gain and loss of many genes across the pathway at once. In these cases, coevolved modules appear to reorganize as units and produce new chemistry in the process (Bird et al., 2025 Genetics; Agosto-Ramos et al., 2025 The Plant Cell). With the phylogenetic resolution to interrogate these gene gain and loss events, I also argued that some metabolic innovations previously attributed to whole-genome duplication may instead reflect more rapid and complex cycles of gene gain and loss (Bird et al. 2025 Current Opinion in Plant Biology).
Where this is going
While much of my future work will remain focused on all three questions above, I also see the longer-term vision of this program as connecting genome restructuring to biodiversity itself and asking whether lineages that restructure their genomes more readily also diversify faster, occupy broader niches, or generate greater phenotypic disparity. Bringing genome architecture together with phylogenetic, ecological, and trait data is an exciting area made tractable with recent advances in genomic sequencing, and it’s a direction I hope to take my work.
“Humane” Genomics
Genetics and evolutionary biology have a uniquely dark history due to their central role in justifying eugenics, colonialism, and racism. Moreover, misunderstandings about human genetics and evolution result in genetic essentialism, the belief that a “race” is a genetically homogenous grouping of people, and that races primarily differ physically, cognitively, and behaviorally because of genetic differences. In turn, genetic essentialism is associated with opposition to policies that promote racial equality.
Recent advances in genomics, like Genome-Wide Association Studies and polygenic scores, and the widespread availability of public databases have fueled a resurgence in scientific racism. Additionally, some mainstream research perpetuates overly simplistic and deterministic ideas about genetics that further facilitate racist misuse. As subject matter experts, there is an obligation for researchers trained in these fields to understand this history, how this history affects contemporary ideas and debates, and how to move the field and public understanding forward.
My work in this area has focused on three main aspects. First, I use methods from genetics and genomics to directly challenge racist hypotheses about genetics and racial differences, especially pertaining to the longstanding theories about IQ scores. Second, I focus on conceptual and methodological problems in mainstream research that contribute to biological determinism and reinforce genetic essentialism. Third, I work to create and implement biology curricula, rooted in pedagogical theory, that teach students the latest in genetics and evolution while explaining how cutting-edge science refutes genetic essentialism. Such curricula have shown strong evidence of reducing racial prejudice in students and may also inoculate them against race science propaganda that is prevalent in some online communities.